Alzheimer-Demenz
Im alltäglichen Sprachgebrauch werden die Begriffe Alzheimer und Demenz häufig gleichbedeutend benutzt. Allerdings ist Alzheimer nur eine Form von Demenz, wenn auch mit 60-70% aller Fälle die häufigste. Daneben gibt es noch über 50 verschiedene andere Demenzformen, wobei die vaskuläre Demenz, die Lewy-Körperchen-Demenz und die frontotemporale Demenz die häufigsten sind.
Allein in Deutschland gibt es über eine Millionen Demenzpatienten – Tendenz steigend. Allen Demenzformen gemeinsam ist der fortschreitende Verlust von Synapsen und Gehirnzellen, die zu starken Beeinträchtigungen von Gedächtnis, Intelligenz und Verhalten.
Die Alzheimer-Demenz selbst wurde 1906 zuerst von Alois Alzheimer beschrieben, der ihr auch den Namen gab.
Krankheitsbild
Bei der Alzheimer-Krankheit verschlechtert sich das Erinnerungs- und Orientierungsvermögen immer mehr. Im Verlauf der Erkrankung treten auch weitere Symptome wie Persönlichkeits- und Verhaltensänderungen auf. In der Regel werden drei Stadien der Erkrankung unterschieden:
Frühes Stadium - leichte Demenz
- Unfähigkeit, Neuinformationen zu speichern
- erst Schwierigkeiten mit neuen Situationen, dann findet sich Patient auch in vertrauten nicht mehr zurecht
- Kurzzeitgedächtnis lässt frühzeitig nach (kann schwierig von altersbedingter Vergesslichkeit zu unterscheiden sein)
- ausgeprägten Merkfähigkeits-, Orientierungs-, Denkstörungen
- Gefühle, Persönlichkeit, äußeres Auftreten bleibt oft lange Zeit intakt
Mittelgradig schwere Demenz
- Hilfsbedürftigkeit im Alltag
- Ablaufreihenfolgen werden vergessen (z.B. Bedienen d. vertrauten Kaffeemaschine, Zuknöpfen d. Hemdes)
- emotionale Instabiltät, Weinkrämpfe, Stimmungsschwankungen, Schwankungen in der Gedächtnisleistung, zwanghafte Verhaltensweisen
Endstadium - schwere Demenz
- vollkommene Abhängigkeit
- Reflexe aus frühester Kindheit (Greif-, Saugreflex)
- nahe Angehörige werden nicht mehr erkannt
- Störung des Tag-Nacht-Rhythmus
- schließlich Bettlägerigkeit
- organische Fkten zunehmend beeinträchtigt, Kontrolle über Blasen- /Darmtätigkeit versagt
- schließlich unfähig zu kauen u. zu schlucken ð Gefahr d. Lungenentzündung
ð häufigste Todesursache Infektionen d. Atemwege
Diagnose
Demenzen wie die Alzheimer-Demenz sind von erfahrenen Ärzten in der Regel recht leicht anhand der Schilderung der typischen Beschwerden (auch durch Angehörige), des Auftretens des Patienten und einer sorgfältigen Untersuchung zu erkennen. Neuropsychologische Tests helfen Gedächtnisdefizite zu quantifizieren. Die am häufigsten verwendeten sind
- der 15-minütige MMSE (Mini Mental State Examination / Mini Mental Status Test)
- die erweiterte Version als 30-minütiger SIDAM (Strukturiertes Interview für die Diagnose einer Demenz vom Alzheimer Typ)
- weitere, schnellere und Schwächen des MMSE ausgleichende Tests wie DemTect (Demenz-Detections-Test) oder TFDD (Test zur Früherkennung von Demenzen mit Depressionsabgrenzung)
Bildgebende Verfahren wie MRT (Magnetresonanztomograph) und CT (Computertomograph) können die Verkleinerung des Gehirns bei Alzheimer-Patienten v.a. im Bereich des Temporal- und des Frontallappens zeigen. Die PET (Positronenemissionstomographie) kann den verminderten Stoffwechsel in diesen Regionen darstellen. Blutuntersuchungen und ggf. auch eine Hirnwasseruntersuchung (Liquorpunktion) können helfen, andere Krankheiten auszuschließen, Denn, da viele Befunde auch auf andere Erkrankungen zutreffen können, ist die Alzheimer-Diagnose immer eine Ausschlussdiagnose. Eine gesicherte Diagnose kann erst nach dem Tod durch eine Untersuchung des erkrankten Gehirns und Nachweis der typischen Veränderungen (siehe unten) erfolgen.
Was passiert im Gehirn bei Alzheimer?
Die Ursachen der Alzheimer-Demenz und die neurophysiologischen Hintergründe sind noch nicht abschließend geklärt.
Der für Demenzen typische Gehirnschwund wird durch ein Schrumpfen der Nervenzellen und einen Verlust von Zellkontakten (Synapsen) hervorgerufen. Dabei gehen ganze Hirnregionen zugrunde, was zu einem Mangel an bestimmten Neurotransmittern, v.a. an Acetylcholin aus dem Nucleus basalis Meynert, führt. Dieser Acetylcholinmangel wird vor allem für die Störung des Gedächtnisses, der Aufmerksamkeit und der Konzentration verantwortlich gemacht. Dieser Mangel geht gleichzeitig und möglicherweise auch ursächlich mit einem Überschuss an Glutamat einher, das wiederum in dieser erhöhten Konzentration giftig auf den NMDA-Rezepter an den glutamatergen Synapsen wirkt (Exzitotoxizität). Schließlich reagieren auch noradrenerge und serotoninerge Systeme mit, wodurch es – häufig schon als erste Symptome überhaupt zu Krankheitsbeginn – zu Verhaltensauffälligkeiten wie Depressionen, Angst und Unruhe kommt.
Im Zusammenhang mit dem Untergang von Nervenzellen stehen krankhafte Proteinbildungen im Gehirn, die Neurofibrillen (Tau-Protein) einerseits und die Amyloid-Plaques (beta-Amyloid) andererseits.
Neurofibrilläre Bündel (Tau-Protein)
Das Tau-Protein ist ein Protein, das spezifisch nur im Nervensystem vorkommt. Es unterstützt die Bildung und Stabilität der Mikrotubuli. Mikrotubuli kann man sich als gerade, hohle Röhrchen mit einem Durchmesser von 25nm und einer Länge von 200nm – 25µm vorstellen, deren Wand aus alpha- und beta-Tubulin-„Kügelchen“ gebildet wird. Sie dienen als Teil des Cytoskeletts der Zelle der Aufrechterhaltung der Zellform, den Zellbewegungen, der Bewegung der Chromosomen bei der Zellteilung und der Bewegung von Organellen und Vesikeln. Die Dynamik und Stabilität der Mikrotubuli muss ständig neuen Transportbedürfnissen angepasst werden, was von Proteinen wie u.a. dem Tau-Protein gesteuert wird. Je nach Erfordernissen binden diese Proteine stärker oder weniger stark an die Mikrotubuli, ein Vorgang, der über die Phosphorylierung (Anheftung von Phosphatgruppen) und Dephosphorylierung (Entfernen von Phosphatgruppen) dieser Proteine gesteuert wird. Im Normalfall binden 1-2 Phosphatreste an das Tau-Protein, um es zu regulieren. Bei der Alzheimer-Krankheit aber wird dieses Tau-Protein hochgradierig phosphoryliert, also mit Phosphatgruppen versehen. Dadurch verliert das Tau-Protein seine Bindung zu den Mikrotubuli, die daraufhin ihre Funktion verlieren und zerfallen. Dadurch werden Stofftransport und Reizweiterleitung in der Nervenzelle unterbrochen. Die hyperphosphorylierten Tau-Proteine lagern sich zu dicht gepackten Proteinsträngen zusammen, wodurch die Transportvorgänge in der Zelle schließlich vollständig blockiert werden. Diese Zusammenlagerungen von Tau-Proteinen werden mikroskopisch als so genannte Neurofibrilläre Bündel sichtbar.
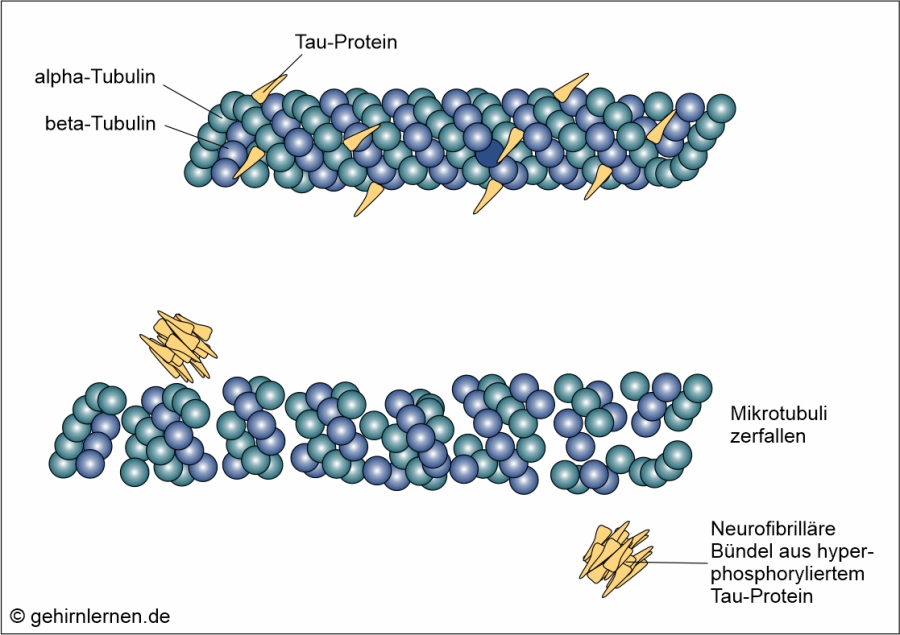 Abb. 14.1: Die Entstehung neurofibrillärer Bündel (oben: Im gesunden Gehirn werden die Mikrotubuli durch Tau-Proteine stabilisiert und kontrolliert; unten: Im Alzheimer-Gehirn lösen sich hyperphosphorylierte Tau-Proteine von den Mikrotubuli und lagern sic
Abb. 14.1: Die Entstehung neurofibrillärer Bündel (oben: Im gesunden Gehirn werden die Mikrotubuli durch Tau-Proteine stabilisiert und kontrolliert; unten: Im Alzheimer-Gehirn lösen sich hyperphosphorylierte Tau-Proteine von den Mikrotubuli und lagern sic
Amyloid-Plaques (beta-Amyloid)
Die zweite mikroskopische Auffälligkeit im Gehirn von Alzheimer-Patienten sind die so genannten Amyloid-Plaques, die 1853 als erstes von Rudolph Virchow beschrieben und benannt wurden. Sie bestehen aus einer zentralen globulären Masse, die von veränderten Neuriten und degenerierten Nervenzellkörpern umgeben ist. Die zentrale Masse besteht aus einem Protein in beta-Faltblattstruktur, dem beta-Amyloid. Dieses entsteht aus dem Amyloid-Vorläuferprotein (beta-amyloid-precursor-protein), kurz APP. APP ist ein Membranprotein, das sowohl in der prä- als auch der postsynaptischen Membran vorhanden ist und die Kommunikation zwischen Nervenzellen kontrolliert. Im gesunden Gehirn wird APP durch alpha-Sekretasen geschnitten. Das entstehende wasserlösliche sAPP übernimmt Funktionen bei der Bildung von Synapsen, ist in Lernprozesse und Gedächtnisbildung involviert. Im Gehirn von Alzheimer-Patienten wird das APP aber nicht durch die alpha-Sekretase, sondern durch beta- und gamma-Sekretasen geschnitten. Die entstehenden Bruchstücke, die als beta-Amyloid bezeichnet werden, lagern sich zu unlöslichen Plaques zusammen, die die Reizweiterleitung blockieren. Da ein solcher Vorgang im gesunden Gehirn eigentlich durch so genannte Chaperone unterbunden würde, muss angenommen werden, dass auch der Abbau von beta-Amyloid durch die Chaperone bei der Alzheimer-Krankheit gestört ist.
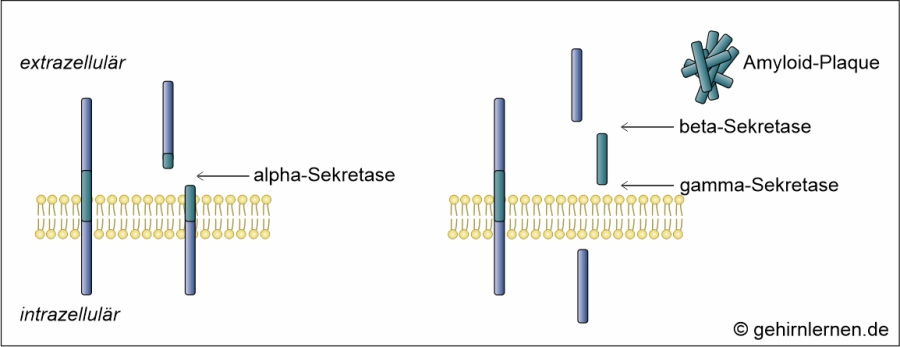 Abb. 14.2: Entstehung von Amyloid-Plaques. (links: Normalerweise wird APP durch die alqha-Sekretase geschnitten; rechts: Wird APP durch die beta- und gamma-Sekretase geschnitten, bilden sich aus den Bruchstücken so genannte Amyloid-Plaques.)
Abb. 14.2: Entstehung von Amyloid-Plaques. (links: Normalerweise wird APP durch die alqha-Sekretase geschnitten; rechts: Wird APP durch die beta- und gamma-Sekretase geschnitten, bilden sich aus den Bruchstücken so genannte Amyloid-Plaques.)
Lange Zeit wurden die unlöslichen Amyloid-Plaques als zentral in der Pathogenese der Alzheimer-Krankheit angesehen. Da Plaques aber auch in normal alternden Gehirnen zu finden sind, wurde der Fokus in den letzten Jahren vermehrt auf das lösliche beta-Amyloid gelegt, dem ein neurotoxisches Potenzial (also eine Giftigkeit für die Nervenzellen) nachgewiesen werden konnte. Dieses scheint Nervenzellen zu töten und auch entscheidenden Einfluss auf das Immunsystem zu nehmen.
Welche Hirnregionen sind betroffen?
Die oben genannten Prozesse der Alzheimer-Erkrankung beginnen im Temporallappen, genauer gesagt im Hippocampus, dem Bereich, der maßgeblich für den Transfer von Gedächtnisinhalten aus dem Kurz- in das Langzeitgedächtnis zuständig ist. Zusätzlich ist das Vorderhirn mit dem Nucleus basalis Meynert betroffen, in dem die Fasern des Neurotransmitters Acetylcholin ihren Ursprung nehmen. Der daraus resultierende Acetylcholin-Mangel trifft wiederum am stärksten den Hippocampus, da dieser den Neurotransmitter für die Gedächtniskonsolidierung benötigt. Diese Beobachtungen korrelieren mit den ersten Symptomen der Alzheimer-Krankheit, nämlich dem Nachlassen des Kurzzeitgedächtnisses, den Problemen beim Speichern neuer Informationen, der Schwierigkeit mit neuen Situationen zurecht zu kommen.
Im Verlauf der Erkrankung sind dann weitere Gehirngebiete insbesondere im Frontal- und Temporallappen betroffen, wodurch zu den Gedächtnisproblemen weitere kognitive, emotionale und soziale Einschränkungen hinzukommen. Im Spätstadium betrifft die Erkrankung dann schließlich das gesamte Gehirn.
Während die Ausbreitung der Neurofibrillen in den genannten Gebieten mit der Stadieneinteilung korreliert, kann die Verteilung der Amyloid-Plaques nicht mit dem Schweregrad der Krankheit in Verbindung gesetzt werden.
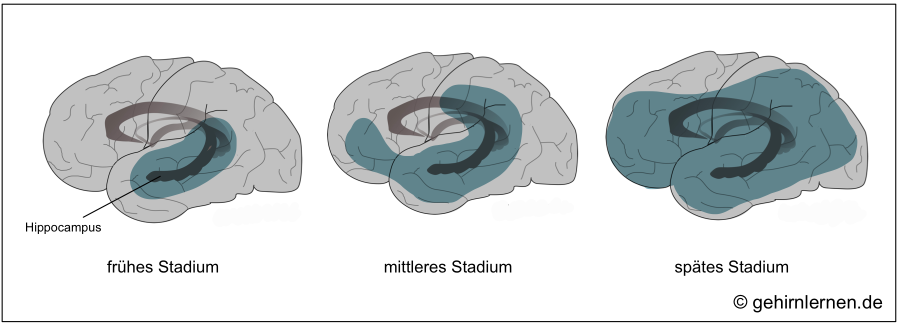 Abb. 14.3: Die Alzheimer-Erkrankung im Verlauf
Abb. 14.3: Die Alzheimer-Erkrankung im Verlauf
Was sind die Ursachen dieser Veränderungen?
Die Ursachen der Alzheimer-Demenz sind nicht vollständig geklärt. Es gibt eine familiäre Form der Alzheimer-Demenz, die autosomal-dominant vererbt wird. Die Betroffenen erkranken sehr früh, im Alter zwischen 30 und 65 Jahren. Betroffen sind hier ein oder mehrere der Gene APP (Chromosom 21), Presenilin-1 (Chromosom 14) und Presenilin-2 (Chromosom 1). Diese genetische Form der Alzheimer-Demenz macht jedoch nur ca. 1 % der Fälle aus.
In den anderen 99 % Fällen ist das Alter der größte Risikofaktor. Ab dem 65. Lebensjahr steigt das Risiko zu erkranken stark an. Zudem scheint eine Mutation im Apolipoprotein Epsilon 4 (ApoE4, Chromosom 19) eine Rolle zu spielen. Diese konnte bislang aber auch nur in 25% der Alzheimer-Patienten nachgewiesen werden. Zudem führt das Vorliegen der Mutation nicht zwangsläufig zur Erkrankung, so dass weiterhin das Alter als wichtigster Risikofaktor angesehen wird. Zudem scheinen ein aktiver Lebensstil mit unterschiedlichen sozialen Kontakten, geistiger Aktivität, regelmäßiger Bewegung und gesunder Ernährung sowie der Verzicht auf Drogen, Alkohol und Nikotin einen gewissen schützenden Effekt auf die Entstehung einer Demenz ausüben zu können.
Welche Therapien gibt es gegen die Alzheimer-Demenz?
Die Alzheimer-Demenz ist nach heutigem Stand der medizinischen Forschung nicht heilbar, ihr Verlauf kann aber positiv beeinflusst werden. Hierzu sind eine frühe Erkennung und Behandlung wichtig. Eine medikamentöse Therapie mit so genannten Antidementiva kann den Verlauf um 1-2 Jahre verzögern. Zum Einsatz kommen vor allem im Frühstadium Acetylcholinesterase-Hemmer. Diese hemmen, wie der Name schon sagt, die Acetylcholinesterase im synaptischen Spalt. Dadurch kann diese Acetylcholin nicht bzw. nur vermindert spalten. So bleibt Acetylcholin länger im synaptischen Spalt aktiv und die Reizweiterleitung über Acetylcholin wird damit verstärkt. Diese können im Verlauf durch Antagonisten am glutamatergen NMDA-Rezeptor ergänzt werden. Ein Wirkstoff hier ist Memantin. Memantin blockiert den Kanal des geöffneten NMDA-Rezeptors, wodurch ein krankhaft erhöhter Calcium-Einstrom, der die Nervenzellen schädig (Exzitotoxizität, s.o.), unterbunden wird. Die kognitiven Leistungen können hierdurch vor allem im mittleren und späten Stadium der Erkrankung verbessert werden.
Neben den medikamentösen Maßnahmen wird die Therapie durch psycho- und sozialtherapeutische Maßnahmen wie Milieu-Therapie, Selbsterhaltungs-Therapie und Gedächtnistraining ergänzt. Die Patienten sollen sich gut aufgehoben fühlen, spüren, dass die verlangsamte Denkweise nicht zum Ausschluss von allen sozialen Aktivitäten führt, um die Lebensqualität möglichst lange zu erhalten.